Vous n’auriez pas vu un gène, par hasard ?
1. Identifier les gènes
Étape préliminaire à toute autre analyse, le séquençage est la détermination, grâce à des méthodes de biologie moléculaire, de l’enchaînement des nucléotides d’une molécule d’ADN.

L’ADN se présente sous la forme d’une double hélice.
© ORNL – U.S. Department of Energy Human Genome Program
Pour reprendre la métaphore la plus couramment employée : il s’agit de retranscrire un texte écrit dans un alphabet de quatre lettres, A, C, G et T (initiales désignant les quatre nucléotides). Dans ce « texte brut », qui dépasse les trois milliards de caractères chez l’être humain, existent des parties codantes, contenant les instructions qui permettent à la machinerie cellulaire de fabriquer les protéines : ce sont les gènes. Or les protéines sont des molécules essentielles au fonctionnement de tous les êtres vivants, dont elles constituent, avec les lipides et les glucides, l’un des trois matériaux de base. On comprend donc aisément l’intérêt que représente la connaissance des séquences des gènes.
Des programmes systématiques
La perspective d’avoir accès à de telles sources d’informations a motivé les programmes de séquençage systématique de génomes. Depuis la fin des années soixante-dix, la taille des génomes étudié est allée crescendo : on a d’abord étudié les génomes d’organismes monocellulaires, en commençant par les plus élémentaires, comme les virus, puis, dans les années quatre-vingt-dix, on s’est intéressé aux bactéries et aux levures. Depuis 1998 ont été étudiés des organismes pluricellulaires (tout d’abord un ver : C. elegans, une mouche : D. melanogaster, la plante A. taliana, bientôt le riz, la souris…).

Crédits photos : INSERM (bactérie, drosophile), Laurent Le Piouff (petit singe), PhotoAlto (fillette), INRA (haricots verts).
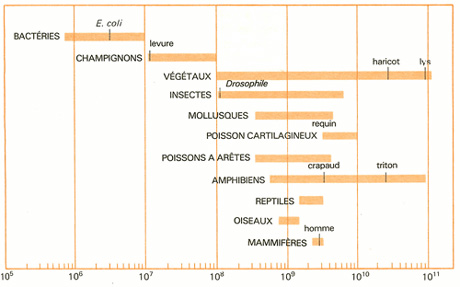
Comparaison du nombre de paires de nucléotides par génome.
Le programme le plus ambitieux et le plus médiatisé est celui portant sur le génome humain, dont une version quasiment « définitive » est à présent disponible. Mais une fois le séquençage effectué, le travail ne fait que commencer.
Apprendre à lire
La tâche est ardue car le fameux « texte » de trois milliards de caractères ne se lit pas d’une traite… loin de là. Et ce n’est pas une spécificité humaine, il en va de même pour les génomes de tous les organismes. Il faut arriver à distinguer les parties porteuses d’information, les parties codantes. En effet, les gènes se trouvent au milieu d’ADN dit non codant, dont la proportion est faible chez les procaryotes mais peut atteindre plus de 98% chez les eucaryotes. Il faut donc identifier, noyés dans un flot de « texte » sans signification apparente, en l’absence de ponctuation et d’espaces, les « mots » que représentent les gènes… Et la difficulté ne s’arrête pas là : il y a en fait non pas une, mais six façons de « lire » la séquence. Ultime complication : les gènes eucaryotes sont morcelés comme les éléments d’une mosaïque.
Cette recherche de gènes, réalisable « manuellement » sur de courts fragments d’ADN, est inconcevable à l’échelle génomique sans l’aide d’outils informatiques, adaptés au génome analysé. Pour davantage d’efficacité, les logiciels employés combinent souvent plusieurs méthodes.
À la recherche d’indices
Une première méthode est basée sur la présence d’indices permettant de localiser les séquences codantes. En effet, celles-ci se terminent par des triplets de nucléotides (codons), appelés « codons STOP », qui sont toujours les mêmes. Entre deux codons STOP se cache donc peut-être une région codante. S’ajoutent à cela, situées en amont du gène éventuel, des séquences caractéristiques sur lesquelles se fixent les enzymes qui « lisent » l’ADN lors de la première étape de fabrication des protéines. Des algorithmes ont été développés afin de traquer ces indices tout au long de la séquence d’ADN.
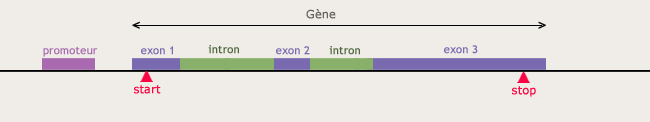
Localisation d’un gène.
De plus, le « style » des séquences codantes, évalué par la mesure des fréquences relatives d’assemblage des quatre lettres, diffère de celui des séquences non codantes. Des outils mathématiques, les plus efficaces étant les modèles de Markov, sont capables de détecter de telles variations de « style » et indiquent les régions de la séquence susceptibles d’être codantes.
À la recherche de ressemblances
Une autre méthode est la recherche des séquences d’ADN similaires à celle qu’on étudie, parmi les millions de séquences stockées dans les diverses banques de données. Ce « criblage » de banques consiste à tenter d’aligner au mieux la séquence étudiée avec celles déjà répertoriées. Un des logiciels de recherche de similarité les plus employés est BLAST, mis au point par le National Center for Biotechnology Information aux États-Unis. Compte tenu de la longueur des séquences d’ADN à comparer, l’utilisation d’algorithmes se révèle très coûteux en temps. C’est pourquoi ce type de logiciel procède selon une démarche heuristique et, sans garantir la solution optimale, propose rapidement plusieurs séquences similaires pertinentes.
Une fois ces différentes données compilées, les résultats ainsi obtenus in silico se révèlent assez fiables lorsqu’ils sont appliqués aux génomes procaryotes, mais beaucoup moins pour les génomes eucaryotes. Dans tous les cas, ce ne sont que des prédictions qui doivent être confirmées expérimentalement par les biologistes.
Mais l’annotation syntaxique de la séquence d’ADN ainsi obtenue n’est que le préalable à une mission encore beaucoup plus ambitieuse : déterminer la fonction des gènes.
2. Rechercher la fonction des gènes
La démarche suivie par les bio-informaticiens pour découvrir la fonction d’un gène « non identifié » est de rechercher, par criblage de banques de données, des gènes de fonction connue ayant une séquence similaire à celle étudiée. Une similarité de séquence reflète souvent l’existence d’un gène ancestral commun et peut se traduire par une fonction analogue, l’homologie de structure appelant une homologie de fonction. On touche ici au domaine de la phylogénétique.
Le précepte de base de la phylogénétique se résume ainsi : tous les génomes dérivent d’un génome ancestral unique, qui a évolué à la suite de mutations successives. Ces mutations sont apparues aléatoirement, et la probabilité qu’elles se produisent est comparable quels que soient l’organisme et le gène. Des gènes homologues provenant de deux espèces proches sur l’arbre du vivant auront donc un degré de similitude supérieur à ceux d’espèces éloignées. La reconstruction d’arbres phylogénétiques est, du reste, un domaine de recherche en bio-informatique à part entière.
Depuis son énoncé, la théorie de Darwin s’est progressivement imposée et il est à présent communément admis que tous les êtres vivants actuels descendent d’un ancêtre commun. Jusqu’aux années soixante, les travaux de classification tenant compte de l’histoire évolutive des espèces étaient limités à l’échelle « macroscopique » : comparer leurs morphologies, leurs comportements et leur distribution géographique. La découverte du support de l’hérédité puis de l’universalité du code génétique, et la disponibilité des séquences d’ADN et de protéines ont, dans ce domaine aussi, ouvert des perspectives nouvelles. C’est au niveau moléculaire que se recherchent à présent les « liens de parenté » unissant les différents organismes.
Pour cette recherche, les outils informatiques permettent une analyse des différents gènes d’un génome entier donné, ou bien l’étude de gènes homologues chez un très grand nombre d’organismes et d’espèces différentes. Dans tous les cas, des algorithmes de comparaison et d’alignement de séquences sont sollicités. Le principe est simple : il s’agit d’aligner plusieurs séquences codantes homologues, la « distance » entre ces séquences (c’est-à-dire leur degré de similitude) est alors proportionnelle à la distance relative des organismes correspondants sur l’arbre de vie.
Bien sûr, cette approche n’exclut pas les méthodes plus anciennes de phylogénie et vient compléter les enseignements apportés par des disciplines comme la paléontologie, l’éthologie ou des études d’embryogenèse.
La génomique montre ses limites
Notons que le type de travaux que nous venons de décrire rencontre parfois des limites dues aux banques de données. Leur nombre élevé et la nature très diverse des informations qu’elles contiennent créent un manque d’homogénéité qui peut nuire à l’automatisation des recherches. Un grand travail serait nécessaire afin de rendre plus cohérentes ces mines d’informations.
Plus important, les différents volets de l’analyse génomique qui viennent d’être décrits ne permettent pas l’identification directe des gènes, mais se bornent à une prédiction de leur structure et de leur fonction. Et surtout, la représentation de la cellule à laquelle donne accès la génomique est « statique », et ne prend en particulier pas en compte le fonctionnement de la cellule au cours du temps. Connaître l’existence de tel gène, à tel endroit du génome, codant pour telles protéines, peut être considéré comme le premier niveau de compréhension. C’est pour compléter cette approche que s’est développée la post-génomique, qui étudie les produits d’expression des gènes : ARN messagers (ARNm) et protéines, et permet d’accéder au niveau supérieur : mieux comprendre le fonctionnement même de la cellule.
Niveau de lecture
Aidez-nous à évaluer le niveau de lecture de ce document.
Votre choix a été pris en compte. Merci d'avoir estimé le niveau de ce document !

